在日常實驗中,我們經常依賴吸光值來估算濃度、追蹤反應變化或判別樣品品質。然而你是否也碰過這種情況:同一組樣品,測得的數值忽高忽低?做完實驗後,畫出的標準曲線卻與預期不符?這些吸光值不穩定的現象,常讓人懷疑樣品是不是已經變質,或是步驟哪裡出了差錯。事實上,往往只是一些容易被忽略的小疏漏在作祟。我們特地整理出一張實用對照表,幫助你在遇到數據飄忽不定時,快速排查問題、縮短 troubleshooting 時間!
💡 一張表快速排查 10 個常見吸光值測量誤差來源
| 常見問題情境 | 可能原因 | 改善建議 |
|---|---|---|
| 同一組樣品,數值忽高忽低 | 樣品混合不均、有沉澱或氣泡 | 測量前以 vortex 混勻樣品,確認無沉澱與氣泡 |
| 空白組或對照組吸光值異常偏高 | 試劑汙染、緩衝液本身吸光 | 更換新的試劑或緩衝液,避免空白組放置過久 |
| 吸光值太高,超過量測範圍 | 樣品濃度太高,光無法穿透 | 適度稀釋樣品或改用短光徑比色皿 |
| 標準曲線不線性,結果失準 | 超出儀器線性範圍或稀釋錯誤 | 控制 OD 在 0.1–1.0 間,重建標準曲線 |
| ELISA 數值差異大,難以判讀 | 加樣量不均、微量盤放置歪斜 | 使用 8 爪或 12 爪 pipette 同步加樣,並確保微量盤放置位置 |
| 數值偏低、不如預期 | 波長設定錯誤或反應時間不足 | 確認波長設定正確,反應終止時間一致 |
| 不同天測值落差大,無法比對 | 光源不穩或儀器未校正 | 定期校正儀器並記錄批次與測定條件 |
| OD600 數值異常或跳動大 | 濃度過高產生光散射效應 | 保持 OD600 在 0.1–0.8,必要時適度稀釋樣品再測量 |
| 酵素動力學反應變化太快,抓不到初速 | 酵素或受質 (Substrate) 濃度過高 | 降低濃度或縮短反應時間,聚焦在初速階段 |
| 使用比色皿後數值變得不穩定 | 比色皿有刮痕或殘留物影響光路 | 每次使用前後確認比色皿無刮痕或指紋 |
💡 吸光度測量原理快讀
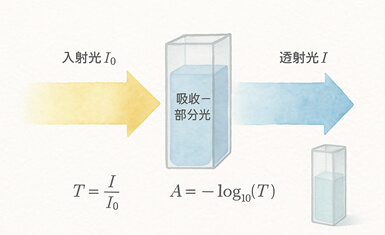
當一束光照向樣品時,一部分的光會被吸收、剩下的光則穿透樣品。我們可以用「透光率(Transmittance,簡寫為 T)」或「透光百分率(%T)」來表示樣品讓多少光通過。T = I / I₀透光率 T 是通過樣品後的透射光強度 I 與入射光強度 I₀ 的比值%T = T x 100透光百分率是透光率乘以 100
為了更清楚表達樣品對特定波長光的吸收能力,通常會將透光率對數轉換為「吸光度(Absorbance,簡寫為 A)」。A = −log₁₀(T) = 2 − log₁₀(%T)吸光度越高,代表樣品吸收的光越多、透光率越低
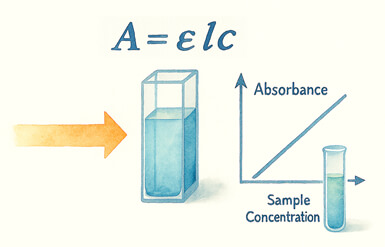
吸光度也常被稱為「吸光值」或「光密度 (Optical density, OD)」。在實驗條件控制得當的情況下(如光源穩定、波長正確、樣品均一),吸光度與樣品濃度會呈現良好的線性關係,因此可以用來估算濃度。其計算原理來自比爾-朗伯定律 (Beer-Lambert Law),常見公式如下:A = εlc其中:
A:吸光度(吸光值、OD)
ε:摩爾吸收係數(與波長、分子性質相關)
l:光徑長度(光穿過樣品的路徑長度,通常為比色皿寬度,常見為 1 cm)
c:樣品濃度(常見單位為 mol/L 或 μg/mL)